...Best of Sicily presents... Best of Sicily Magazine. ... Dedicated to Sicilian art, culture, history, people, places and all things Sicilian. |
by Vincenzo Mormino | ||
Magazine Index Best of Sicily Arts & Culture Fashion Food & Wine History & Society About Us Travel Faqs Contact Map of Sicily |
When referring generically to "thalassemia," the conditions classified as thalassemia major are usually what is referred to. There are fewer than five hundred clinical thalassemia cases in Sicily, in a population of nearly six million. (This number is comparable to the number of Sicilian cases of another blood disease, sickle cell anemia, in which the cellular condition may be quite similar to that of sickle beta thalassemia.) However, estimates of the number of genetic carriers who could transmit thalassemia to their children range from six to twelve percent. This places the statistical frequency of the gene in Sicily between that of Sardinia, where it is more common, and Greece, where it is slightly less frequent. Few Sicilians know anybody who has ever been hospitalized for thalassemia. However, the fact that thalassemia cases are numerically rare in Sicily (affecting far less than one percent of the population) does not negate their high statistical significance, or the wide distribution of the genes which transmit the condition. That said, there is evidence to suggest that the Sicilian frequency of the hereditary trait ("carriers") has sometimes been overstated --though perhaps not intentionally. Regular blood tranfusions are the usual treatment for those suffering from severe symptoms of thalassemia. The problem with transfusions is that the resulting increase of iron levels can create conditions detrimental to various organs. There is no cure, though various therapies are under development to treat the disease, considered the world's most common hereditary single-gene disorder. In most general terms, there are two forms of thalassemia --the actual illness (thalassemia major) inherited through two parents and the less severe anemic state (thalassemia minor) sometimes inherited from one parent. The wide range of severe conditions broadly defined as thalassemia major (and inherited recessively through both parents) are often fatal, claiming their victims before middle age. This
article is not presented as a scientific reference, but a few pragmatic
observations are appropriate. As we've mentioned, there are various forms
of thalassemia. In general terms, beta thalassemia is a condition
that results in an excess of alpha globins, while alpha thalassemia
is a condition resulting in an excess of beta globins. If both parents are
"carriers" of one of the various thalassemia traits, there is
a one in four (25%) probability (not only generally but with each pregnancy) that any child of
the couple will be affected Sardinia, Sicily and Calabria have more cases of thalassemia than other regions of Italy. While Sicily has a complex genetic environment, thalassemia now occurs around the world, having found its way into numerous New World populations. Historically, it was relatively frequent among Italians, Greeks, Turks and Arabs, as well as Persians, Indians and peoples of the Far East. (The map shows Old World distribution.) The disease was not widely identified by its present name until the 1930s, through the efforts of Thomas Cooley and others. Ancient and medieval literature allude to conditions which appear to have been caused by thalassemia, but infant mortality was so high (from this and other causes) and so little known about hemolytic disorders that the true causes of the thalassemic diseases were only determined in the twentieth century, with a greater knowledge of genetics. It is predicted that thalassemia will become a more widespread public health issue in the decades to come. "Thalassemia trait" (the carrier state which facilitates one's transmitting the disease to children) may cause red blood cells to be smaller in size than normal, but there exists no scientific evidence that thalassemia trait itself causes health problems. Some individuals with thalassemia trait have a degree of protection from malaria, a characteristic also identified with sickle cell anemia. (The protective mutations probably occurred many thousands of years ago, the percentage of affected individuals increasing through natural selection as persons vulnerable to malaria were less likely to survive.) This may partially explain thalassemia's distribution in regions prone to malaria and among populations whose very survival may have depended on resistance to malaria. While thalassemia trait is found in many populations, it was historically more frequent in people from regions where malaria is (or was) commonplace. Often displayed by carriers, thalassemia minor is a form of anemia that cannot be treated as a normal iron deficiency but usually presents no serious health risk. In effect, thalassemia minor is a condition which may be inherited from one parent, but the more severe thalassemia major may only be inherited from both parents. The continuing battle against genetic diseases is a constant challenge, with tangible progress measured not in months or even years but in decades and lifetimes, as well as lives. The life of the typical thalassemia patient, wherever he or she may be, depends on the success of the most advanced medical research. A number of organizations around the world support research (and treatment) for thalassemia and related hereditary diseases. One of the more complete and informative websites is published by the Cooley's Anemia Foundation, based in the United States. About the Author: Vincenzo Mormino has written about scientific topics, wildlife and nature for Best of Sicily and hard-copy publications. | |
Top of Page |
 Thalassemia (from the Greek "thalassa" meaning "sea") is a diverse
group of hereditary blood diseases that usually results in the reduced or
flawed production of hemoglobin, the essential respiratory element of red
blood cells. Some cases of thalassemia (or thalassaemia) are debilitating, requiring extensive
medical treatment to keep patients alive. Others require little or no medical
attention. Thalassemia major, a serious
form of the disease, is also known as Cooley's Anemia, for American
physician Thomas Cooley (who resented the disease being named for him), or "Mediterranean Anemia," though it
is present in Asian and African regions far from the Mediterranean basin.
Thalassemia has been present in Sicily for many centuries, probably
since circa 600 BC (BCE), if not earlier. Inherited through a recessive
gene, thalassemia is frequent in the Middle East and the eastern Mediterranean,
and the gene probably arrived in Sicily with the island's extensive colonization
by Greeks and Phoenicians. It is possible that thalassemia existed among
Sicily's native populations --the Elymians, Sicanians and Sicels-- before that
time, as it is postulated that some of these earlier inhabitants migrated
to Sicily from the eastern Mediterranean, albeit probably before 4000 BC. Theoretically, it is possible that some of the very earliest Sicilians developed thalassemia through genetic mutations as protection against malaria many thousands of years ago. (This is discussed later.)
A further historical influx of the thalassemia gene may be associated with
mass immigration of the Saracens (Arabs) in the ninth and tenth centuries,
as this population had a strong Middle-eastern Semitic component.
Thalassemia (from the Greek "thalassa" meaning "sea") is a diverse
group of hereditary blood diseases that usually results in the reduced or
flawed production of hemoglobin, the essential respiratory element of red
blood cells. Some cases of thalassemia (or thalassaemia) are debilitating, requiring extensive
medical treatment to keep patients alive. Others require little or no medical
attention. Thalassemia major, a serious
form of the disease, is also known as Cooley's Anemia, for American
physician Thomas Cooley (who resented the disease being named for him), or "Mediterranean Anemia," though it
is present in Asian and African regions far from the Mediterranean basin.
Thalassemia has been present in Sicily for many centuries, probably
since circa 600 BC (BCE), if not earlier. Inherited through a recessive
gene, thalassemia is frequent in the Middle East and the eastern Mediterranean,
and the gene probably arrived in Sicily with the island's extensive colonization
by Greeks and Phoenicians. It is possible that thalassemia existed among
Sicily's native populations --the Elymians, Sicanians and Sicels-- before that
time, as it is postulated that some of these earlier inhabitants migrated
to Sicily from the eastern Mediterranean, albeit probably before 4000 BC. Theoretically, it is possible that some of the very earliest Sicilians developed thalassemia through genetic mutations as protection against malaria many thousands of years ago. (This is discussed later.)
A further historical influx of the thalassemia gene may be associated with
mass immigration of the Saracens (Arabs) in the ninth and tenth centuries,
as this population had a strong Middle-eastern Semitic component.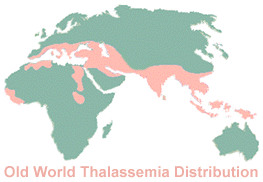 with
the disease, which may take the forms E beta thalassemia, beta
sickle thalassemia, hemoglobin H disease or hydrops fetalis
(or alpha thalassemia major). A complete blood count or iron study
may provide the initial indication of thalassemia. Beta thalassemia trait
(for a "carrier" who might transmit the disease) can be determined
by a simple hemoglobin electrophoresis test. Alpha thalassemia trait
may be identified through the more complex alpha globin DNA mutation analysis.
with
the disease, which may take the forms E beta thalassemia, beta
sickle thalassemia, hemoglobin H disease or hydrops fetalis
(or alpha thalassemia major). A complete blood count or iron study
may provide the initial indication of thalassemia. Beta thalassemia trait
(for a "carrier" who might transmit the disease) can be determined
by a simple hemoglobin electrophoresis test. Alpha thalassemia trait
may be identified through the more complex alpha globin DNA mutation analysis.